脓疱蔓延全身 疼痛难忍反覆发作 GPP非一般牛皮癣 严重恐器衰
医句话:
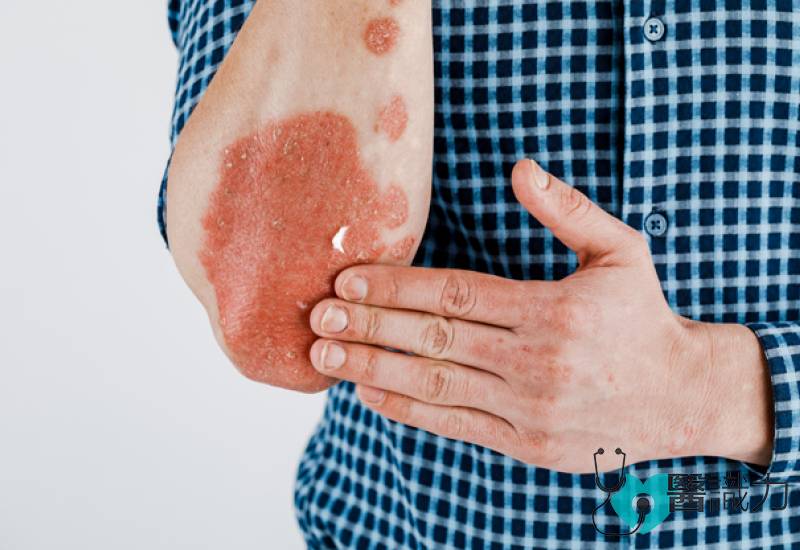
泛发性脓疱型牛皮癣(General Pustular Psoriasis,GPP)是最罕见,却也是最严重的牛皮癣之一。患者会长出布满全身,且疼痛的脓疱,严重者可并发败血症、多重器官衰竭等,危及生命。
长久以来,医学界把其它牛皮癣的治疗方法套用于GPP,但效果有限且缓慢。目前一种针对GPP发病机制的生物制剂问世,成为首个GPP的专治药物,相信能为患者带来裨益。
医句话:
泛发性脓疱型牛皮癣(General Pustular Psoriasis,GPP)是最罕见,却也是最严重的牛皮癣之一。患者会长出布满全身,且疼痛的脓疱,严重者可并发败血症、多重器官衰竭等,危及生命。
长久以来,医学界把其它牛皮癣的治疗方法套用于GPP,但效果有限且缓慢。目前一种针对GPP发病机制的生物制剂问世,成为首个GPP的专治药物,相信能为患者带来裨益。
医句话:
过去的10至15年对遗传学科而言是关键时刻,皆因在这段期间,除了遗传学科医生的临床经验获得提升,其他专科医生也开始对罕见疾病有所认识,不仅主动向遗传学科医生咨询如何辨识罕见疾病患者,并且也会视情况安排基因检测,一旦怀疑为罕见疾病后即转介给遗传学科医生接手。
如此跨学科合作,使得罕见疾病的诊断速度获得提升,让患者能够更早接受适当的治疗,对患者不啻是迟来的福音。
医句话:
过去医生要找出患者的病因是通过几个步骤,分别是问诊、了解家族病史、各种筛检扫描及检查等,通过相关报告来进一步推断患者可能的病症,之后再针对性进行检测,在反复数次后才能做出诊断。不过在面对罕见疾病时,这类做法显得格格不入且费时,往往患者会因此错过治疗黄金时机。得利于科技的进步,如今仅通过基因组检测即可检查患者体内2万个DNA,这也让医生能更快找到患者的病因,包括罕见疾病的存在。
“在众多罕见疾病(rare disease)中,属于先天性代谢缺陷疾病(Inborn Errors of Metabolism,IEM)占了30%,比如枫糖尿症(maple syrup urine disease)、尿素循环障碍(urea cycle disorders)或苯丙酮尿症(phenylketonurua)。其他则属于单基因疾病,例如俗称脆骨症或玻璃娃娃的先天性成骨不全症(Osteogenesis Imperfecta,OI)或马凡氏症候群(Marfan syndrome)等。
医句话:
还记得2022年上映的港剧《白色强人2》吗?剧中主角们极力推动政府全面开放药品清单草案,期望让每位罕见疾病患者获得平等的治疗机会,包括所需的药物。剧情是否曾让你感动?事实上,这部剧所传达的理念,正与今年国际罕见疾病日的主题“公平”相呼应,即希望每位罕见疾病患者都能享有平等权利,获取所需的医疗服务。
“每年2月的最后一天是国际罕见疾病日(Rare Disease Day),今年的主题为‘公平’(equity)。将医疗资源平均分配给所有患者,符合‘平等’(equality)的原则;真正的公平(equity)则是根据患者的健康需求来分配资源,确保每位患者获得所需的医疗服务,包括药物。
公平比平等更重要
举个例子,如果有几个身高不同的人要观看篮球比赛,但是前面有阻碍物,基于平等的原理,每个人都获得同样高度的凳子,但矮个子仍然无法看到比赛。基于公平,每个人将获得不同高度的凳子,确保所有人都能看到比赛。因此,平等是形式上的一致,而公平则是实质上的合理分配,确保所有人都能真正获得相同的机会和结果。
医句话:
天使症候群是一种基因缺陷所致的神经发育障碍疾病,主要表现特征包括学习障碍、运动和平衡障碍、言语缺陷、精神运动迟缓,以及不恰当的拍手大笑等。除了针对天使症候群的症状进行治疗,父母还需要了解这种疾病,接纳孩子的特殊状况,并培养患童的自理能力,让他们在照护中感受到关爱。
“天使症候群(Angelman Syndrome),又称快乐木偶症候群(Happy Puppet Syndrome),是一种基因缺陷引起的神经发育障碍疾病,属于非进展性脑病变。其主要成因是第15条染色体的UBE3A基因异常。虽然大部分原发性自闭症(primary autism)无法找出病因,但天使症候群作为继发性自闭症(secondary autism)的一种类型,其病因可明确追溯至UBE3A基因异常。
在出生时,基因由父母各继承一半,其中UBE3A基因对大脑及神经发育至关重要。如果婴儿从母亲遗传的UBE3A基因出现异常,便可能导致天使症候群。
UBE3A基因异常为致病因
医句话:
血友病的药物不断在进步和创新,让患者能更好地控制出血,保护关节,同时拥有自主生活。治疗方案可分成凝血因子补充疗法、非凝血因子的创新疗法,以及基因疗法3大类。虽然基因疗法已获得成功,但只提供18岁以上的患者,而且费用高昂,且尚未获得我国批准实行。
“血友病主要成因是凝血因子不足,因此治疗方式在于补充缺乏的凝血因子,主要分为需求性治疗或按需治疗(on-demand therapy)和预防性治疗(prophylaxis),前者是针对紧急出血的治疗;后者是定期施予固定剂量的凝血因子,以提高并维持体内凝血因子在一定的浓度,以预防自发性出血长期积累造成的伤害。
血友病患需尽早开始药物治疗,因病情管理的原则在于预防。当然,所谓的‘及早’并非一出生即用药,因为婴儿活动能力有限,出血风险相对不那么高。
医句话:
虽然外观无法发现关节出血,但父母可以观察孩子的动静。关节和日常活动息息相关,本来可以握住玩具的小孩,忽然不愿意用手碰东西了;或小孩不愿意用脚踏在地板、害怕走路或不愿意爬动,那就需要怀疑是不是关节里有些问题。若是等到关节肿胀,那就事态严重了。
“血友病(hemophilia)是一种先天性的出血性疾病,主要症状为凝血功能异常,不容易形成血块来止血。原因是患者血液中缺乏某一种凝血因子(coagulation factor),譬如A型血友病缺乏第八凝血因子;B型血友病则是缺乏第九凝血因子。
凝血因子是参与血液凝固过程的蛋白质,主要有13种。只要缺乏其中一种,就难以形成血栓‘补塞’血管上的破洞以止血。这就像多米诺骨牌效应,每个因子需要发挥各自的作用,相互配合才能顺利地执行止血功能。
其中,A型血友病会比B型更为常见,二者比例约为4:1;至于C型血友病较少见,病情也相对轻微。不过,血友病的轻重并不是根据类型区分,而是血液中凝血因子浓度的高低。
低凝血因子 出血时间更长
医句话:
在马来西亚,每4000人会有1人患有罕见疾病,可能有多达100万名马来西亚人受到一种罕见疾病的影响,大约70%是从童年期开始病发,这也意味着孩童的死亡因素已不再是我们所知道的肺炎、肠胃炎、疟疾、骨痛热症、COVID-19而已。
“罕见疾病有6000种,在欧盟,罕见疾病被定义为每2000人中有1人患上;在美国,该疾病在总人口中的发病人数必须少于20万人。在马来西亚,我们采用了四千分之一的比例,目前世界上有3亿人患有罕见疾病。
简而言之,这意味着虽然个别情况很少见,但总的来说,有成千上万的马来西亚人受到罕见疾病的影响。基本上,他们就像你和我一样,也有基本需求,也需要像其他人一样得到同样治疗的待遇。统计数据显示,它影响了约3.5%至5.9%的人口,这是一个庞大的数字。
医句话:
法布瑞氏症是指在遗传因素下导致人体缺乏了A型α半乳糖甘酶,在这影响下病患会诱发肾衰、心衰、脑血管疾病或神经病变。病患在确诊后除了要接受ERT治疗,家庭成员更需要做基因筛检以提高对这种先天性疾病的意识,病患家庭更可透过产前检查、IVF与PGT,以避免将先天性疾病遗传给下一代。
医句话:
亨廷顿舞蹈症来自于父母遗传(genetic disorder),惟其中10%的病例是因新产生的基因突变所导致。该疾病是一种遗传性的神经退化疾病,会导致脑细胞死亡,通常在30岁至50岁之间病发。
“亨廷顿舞蹈症(Huntington's Disease,HD),也称亨廷顿病是一种遗传性的神经退化疾病,会导致脑细胞死亡,发病年龄介于30岁至50岁,但也可以在任何年龄开始发病。
在带有与罹患此症相关异常的染色体家族中,相关成员可能于较年轻时就发病。大约8%的病例在20岁以前发病,典型症状与帕金森氏症相似,此症患者经常会低估了自身病情。
在全球,每1万人中就有1人患有此疾病。在欧裔人士中,此症的盛行率为每10万人中4至15人。在马来西亚里,每1万人,只有0.0024人(不到1人)患有此病,从中得知,此疾病在马来西亚相当罕见,在亚洲也是如此,而目前还没有非洲人口的盛行率数据。此症的病发比例,男性及女性相近。